Examining the structure of the ANSI and ISO usability standards is the first step to developing medical devices that protect users from accidents.
If you want to learn about usability, then reading a standards document may not be the easiest starting point. However, standards that address usability for medical devices do exist. Companies must be familiar with these requirements if they want to prove to FDA or other regulatory bodies that they have a usability program in place. These standards also provide information that is specific to the medical industry and is not readily available in more general usability texts.
One way to get started is to look at the structure of the standards and how they relate to developing usable medical devices. The two standards that most drive the requirements in this area are ANSI/AAMI HE74:2001 and IEC 60601-1-6 (referred to here as the standards).1,2 These two standards are very similar to each other. However, there are differences between the standards that must be recognized.
Use Error [sic]
If FDA regulates your company and your products, there is good reason to observe good practices in the area of usability. There are many techniques that are broadly accepted in industry as contributing to better usability. The literature that describes these techniques provides the basis for usability as an engineering discipline. These techniques are summarized in the standards.
More than one-third of medical device incidents reported to FDA involve use error.3 It is significant that FDA documentation and the standards refer to "use error" rather than the more colloquial "user error." User error means that the user made a mistake, while in fact many accidents happen because users performed actions that seemed perfectly reasonable.
The term use error can be used to describe a larger family of problems, in which incidents could have been avoided had the users taken different actions, but the actions taken may have occurred because of bad usability designs. If a use error occurs repeatedly by different users, the true cause of the problem may lie in the design rather than with the users.
If an incorrect user action can be predicted, the design should be altered to mitigate that hazard. For example, if there is an on-off button on the front panel near other commonly used buttons, it is reasonable to assume that users will occasionally press the on-off button when they intend to press a different one. Accidentally turning off a medical device can have serious consequences. If the button was pressed when someone bumped into the machine, he or she may not notice that the machine turned off. Restarting the device may involve a start-up sequence, and during that time the patient may not receive treatment. And some devices need to be disconnected from the patient during the start-up sequence, leading to further delay.
Since this problem can be foreseen, it should be designed out. If the action cannot be prevented, then an indication that the action occurred may be a good alternative. Some devices beep loudly for a few seconds after they have been turned off. This sound alerts users if they have accidentally turned off the device. It also alerts nearby staff, which is useful if, for example, a visiting child turns off the device.
Reasonably Roreseeable Misuse
Accidentally turning off a device is an error that the designers could predict, and therefore they have the responsibility to mitigate this use error. There is another form of predictable use error defined in the standards: reasonably foreseeable misuse or off-label use. This term applies when a device is used for a purpose other than the one stated by the manufacturer.
Designers have an obligation to mitigate reasonably foreseeable misuse. For example, an adult ventilator may be used on a pediatric patient if no other ventilator is available. Although this scenario may not match the designers' target patient group, the designers should consider potential hazards from this use. In cases where off-label use creates a hazard, sometimes such use could be prevented by the design. However, the standards suggest that off-label use should not be explicitly prevented in the case where no hazard will result from that off-label use.
It is not necessary for a device to behave perfectly when subjected to reasonably foreseeable misuse, but it is necessary that such misuses be considered as part of the design process. They can be considered during task analysis, and they can be considered as items in the failure modes and effects analysis (FMEA), which is part of a product's risk analysis.
Abnormal Use
One category of actions that do not have to be tolerated by the designer is abnormal use. Examples given in an annex to IEC 60601-1-6 include a user deliberately disconnecting the alarm system, thereby preventing the system from alerting users to dangerous conditions. Other examples include the use of an x-ray machine after its overheating indicator has lit, and use of a device after a warning is issued from the device that recalibration is required.
The distinction between abnormal use and reasonably foreseeable misuse is sometimes subtle. One definition is that an action taken by many users can be considered reasonably foreseeable misuse, while an action that is an isolated incident may constitute abnormal use. Unfortunately, this definition can only be applied after the device has had sufficient use to allow measurement of the frequency of these incidents.
It may seem like engineers need a crystal ball to see into the future. In practice, though, the reasonably foreseeable misuse of one device may be similar to that of other devices on the market. If so, the designers can see the misuse in the current market and reasonably predict that the same events will occur with the new design. For example, noninvasive ventilators are often connected to nebulizers to deliver medication to a patient. Many noninvasive ventilators make no claims for the suitability of a nebulizer, so this is off-label use of the device. But because this activity happens in the market, it is reasonable to assume that the same thing will happen on any new device brought to market. Whether the designers should put measures in place to prevent this use of the product depends on whether using the nebulizer in this way could create a hazard.
Icons
The usability standards do not give clear guidance on the use of icons. However, for certain classes of devices, FDA insists on the use of English text to avoid ambiguous icons.
Whether icons without supporting text are acceptable for a device may depend on its application and audience. FDA's guidance document on patient labeling, which applies to labels that must be understood by patients and their families, states that icons may be used only with accompanying text.4
Although the standards do not give explicit guidance on icons, they use icons in some examples of use error. Testing icons independently can be a valuable part of a usability program.
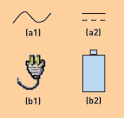
Figure 1. Icon alternatives for technical vs. non-technical users.
For example, engineers might recognize that icons a1 and a2 in Figure 1 represent ac and dc power, respectively. However, non-technical users would probably not understand them. In that case, b1 and b2 might prove to be better alternatives.
Alarms
The usability standards do not address alarms directly, but they do refer to IEC 60601-1-8, which details the alarm requirements.5 One usability challenge often cited in the standards is users setting alarm thresholds too high or too low, effectively disabling the alarm. This problem can be mitigated if the alarm threshold is constantly visible to the user.
IEC 60601-1-8 defines the pattern of alarm sounds and the flashing rate and colors of visual alarm indicators. In that regard, it supersedes BS EN 475, which to date has been the most widely adopted standard for alarm enunciation on medical devices.6
ISO 60601-1 also describes the mechanisms by which alarms can be silenced or disabled. In addition, it suggests icons for use in resetting, silencing, or disabling alarms (see Figure 2).
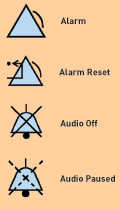
Figure 2. These icons are suggested, but not mandated, by IEC 60601-1-8.
User Documentation
The standards say that the user documentation should be tested as part of the usability testing. Although there is no guarantee that all users will read the user manual, the text must be written in clear prose that makes sense to the user, not just to the design engineers.
One test establishes how often a user requires the user manual to complete a task. In the test, users are told that the documentation is available if they wish to use it. The test records how often the documentation is used, which sections are read, and whether users find the information they sought.
In a separate test activity, users are instructed to consult the documentation to perform a certain task. This type of testing can help ensure that the trial participant uses all sections of the manual.
Usability Process
IEC 60601-1-6 dictates that the documents describing the usability process and the reports from any usability testing be contained in, or referred to from, the usability file. The usability file is the starting point for any investigation of the usability process. From an engineer's point of view, the most important output of a usability program is a better product. However, for FDA, traceability is key. For its inspectors, the critical output is written proof that a well-defined process was followed. A good usability file provides that proof.
Although the standards suggest many established techniques, they do not mandate any particular steps to achieve usability. Therefore, the design team can choose the usability techniques that suit the scale and complexity of their product. However, the standards mandate that a usability specification and a usability validation plan exist.
The usability specification contains details about the target user population and the target patient population. It also includes information about the activities carried out using the equipment, known as the task analysis. The task analysis contains a list of usability hazards with which the device may have to contend. Some portions of this information may consist of references to the product requirements or hazards analysis, which are documents that exist outside of the usability program.
In some cases, the usability specification also includes a style guide to ensure consistency across the interface. For example, on a graphical user interface, it might indicate that all menus must include a title at the top.
The validation plan describes the tests that will establish whether the usability specification has been met. It also lists the usability trials and reviews of the user interface.
By providing documents to embody the requirements and validation, the usability process becomes traceable, which is vital when showing FDA that a process exists and is being followed. Creating these documents as part of the standard infrastructure of every project gives the usability process equal standing with other engineering considerations.
One of the most common failings of a usability process is that it is not owned by any one engineer or group. In some situations, team members may be aware of the shortcomings but not feel obligated to tackle the issue. If no team members take ownership, problems may remain unaddressed. The documents that describe the usability goals and tests provide a focal point for usability issues so that ownership can be assigned.
But the usability process does not stop when a product is launched. Post-market surveillance is an important tool that provides feedback for the next version of the product. Discovering the many ways a product gets used in the field can enable a company to capture market share with next-generation products.
Usability Validation
Validation tests should be based on how the finished device works in a real-world environment, such as a clinical trial. Alternatively, tests can use a prototype of the device and simulate a real-world environment. However, there is a trade-off between these two approaches. Real-world use will expose problems that were missed, because simulations cannot replicate all details of the user environment. On the other hand, prototypes and simulations can be tested much earlier in the design cycle, so problems can be fixed earlier and with a lower cost. This trade-off applies to any usability program, but the standards point out some interesting distinctions between clinical trials and usability testing for medical products.
Clinical Trials. Clinical trials demonstrate safety and effectiveness when the device is used exactly as directed. During trials, the emphasis may be on getting data about the effectiveness of the treatment and on getting efficacy data about the patients being treated. So a usability problem may be resolved with the quickest possible fix to allow the trial to continue, reducing the chance that a more fundamental solution will be addressed later. A trial dedicated solely to usability will not have to juggle these conflicting priorities.
Clinical trials often compromise or prohibit common usability techniques such as photography and video recording. In such trials, the sensitive social contexts of hospitals and privacy concerns are very important.
Usability Testing. IEC 60601-1-6 states that high-risk situations are difficult to investigate in a clinical environment. An event that might happen once per month per device may not occur at all during a two-week trial. If the high-risk, low-probability event does happen, the priority will be on protecting the patient rather than on analyzing the interface. Doing so often means removing the patient from the device under test as the first step. Although this is the safest route, it does not help gather information regarding how the interface could be improved.
Analyzing high-risk, low-probability events requires a different approach from a clinical trial with a real patient. There are two options. FMEA can be applied to user actions in the same way it is used to analyze electrical or mechanical failures. The second approach is to create simulations in which trial users are presented with a mock-up of the scenario that requires investigation. With no patient present, there is no risk involved in introducing equipment malfunctions or alarm conditions.
When trial participants are providing feedback, it is important to observe their actions. Do not be satisfied with their responses to questions about their preferences. The interface that a user says he or she likes is not necessarily the more usable interface. This is especially true in the medical environment. For example, a user may like a device that makes it very easy to disable all alarms—but such a design may compromise safety. Objective measures, such as the time it takes to complete tasks and a user' error rate, provide more scientific validation of the user interface than do questionnaires. The standards point out that users will often unintentionally say they perform a task in a certain way, when in reality they do something different. Therefore, listening to users must be complemented by observation.
Although the standards do not directly address whether trials in multiple countries are necessary, it is a concern for many devices. For example, in the United States, ventilators are typically used by respiratory therapists; that specialization does not exist in Europe. The training and duties of typical users would therefore be different in those two markets. Translations of user manuals and translations of graphical screens or off-screen labels are all subject to testing according to FDA's guidance on labeling.7
Risk Management
Medical usability differs from general usability in the context of risk management. Although usability improvements make the device more pleasant to use and quicker to learn, in a medical device they also reduce risk to the patient. IEC 60601-1-6 refers to risk analysis numerous times and notes that the usability file may be part of the risk management file.
The risk management standard, ISO 14971, requires that use error and reasonably foreseeable misuse be addressed.8 So from a standards compliance stance, conforming to ISO 14971 requires a usability program to be in place.
Medical Device Standards
AAMI HE74 and IEC 60601-1-6 are very closely linked. AAMI cites IEC 60601-1-6 as an equivalent standard to AAMI HE74, and IEC 60601-1-6 contains an informative annex derived from AAMI HE74.
Compared with standards in other industries, these two standards contain a large body of informative guidance. Interestingly, only a small percentage of the guidance is mandatory. The amount of usability work required for a medical device can vary widely, since some user interfaces comprise a single button and others are more complex than a desktop PC.
AAMI HE74 is less strict than IEC 60601-1-6 in defining the exact documents required to support the usability process, leaving product developers more flexibility in how they manage their process. AAMI HE74 also contains an annex that identifies the sections of the FDA quality system regulation that refer to usability.
IEC 60601-1-6 is not an easy standard to use. The table of contents explains very little of what the document contains. One 60-page section gets only one line in the table of contents, the same amount afforded another section that contains two sentences. The document is published as a French and English version, in which all odd pages are in English and all even pages are in French, making it difficult to scroll through a soft copy. The numbering scheme is tied to the other IEC standards, and so, like many usability difficulties, problems are caused by legacy issues. The precedent set by older documents reduces the formatting options available to this document, because the authors must be consistent with previous standards.
In spite of its shortcomings, the IEC document contains useful advice and has enough examples to help companies ascertain what the standard means without having to parse it like a legal contract. Examples include FDA incident reports that illustrate how a better human factors program might have avoided a particular design failure.
Another IEC standard, IEC/CD 62366, "Medical Devices—Application of Usability Engineering to Medical Devices," is currently under development as of this writing. IEC 60601-1-6 was written for electromechanical devices, and the goal of IEC/CD 62366 is to extend that standard to address all medical devices. It is still in draft form, but it will become a significant standard once it has been validated.
Usability Standards Compliance
The increasing complexity of medical equipment means that a well-defined usability program is necessary to protect users and patients from accidents. Usability is becoming more of a science and less of an art. It is increasingly recognized that human factors failures can be eliminated if a process is in place to manage the usability properties of a device. Standards compliance can help companies maintain consistency across multiple projects and can also provide companies with a common language to express their approach to regulatory bodies.
Endnotes
1. AAMI HE74:2001, "Human Factors Design Process for Medical Devices." Washington, DC: Association for the Advancement of Medical Instrumentation, 2001. [back]
2. IEC 60601-1-6, "Collateral Standard: Usability." Geneva: International Electrotechnical Commission, 2004. [back]
3. Patterson, Patricia. "Fitting Human Factors in the Product Development Process." Medical Device & Diagnostic Industry, Vol. 28, No. 1, 2006, pp124-133. [back]
4. "Guidance on Medical Device Patient Labeling; Final Guidance for Industry and FDA". Rockville, MD: U.S. Department of Health and Human Services, Food and Drug Administration, Center for Devices and Radiological Health, 2001. [back]
5. IEC 60601-1-8, "Collateral Standard: Alarm Systems—General Requirements, Tests, and Guidance for Alarm Systems in Medical Electrical Equipment and Medical Electrical Systems". Geneva: International Electrotechnical Commission, 2003. [back]
6. BS EN 475:1995, "Medical Devices—Electrically Generated Alarm Signals". Brussels: European Committee for Electromechanical Standardization, 1995. [back]
7. "Guidance on Medical Device Patient Labeling; Final Guidance for Industry and FDA". Op. cit. [back]
8. ISO 14971:2000, "Medical Devices—Application of Risk Management to Medical Devices." Geneva: International Organization for Standardization, 2003. [back]
This article was published at Medical Device Link in May 2006. If you wish to cite the article in your own work, you may find the following MLA-style information helpful:
Murphy, Niall. "Understanding Usability Standards for Medical Devices," Medical Device & Diagnostic Industry, May 2006.